INSTRUCTIONS
FOR DAY 1 (MASTANI, 2014), EXERCISE 2
Note that the
following color code has been used in this instruction sheet:
Broad headings
are in red.
File names are
in magenta,
Phrases to be
typed into the command line are in blue.
Input
parameters are in dark green.
In this
exercise, we will examine one of the 'post-processing' that you can do once you
have run your scf calculations.
In the
previous exercise, there was only one output quantity that we worked with...
the total energy. Here we will learn to plot the band structure of materials.
We will continue to work with Si,
which was the subject of exercise1.
- STEP1: Go to the
directory ~/hands_on/week1/day1/exercise2 (if you are not already there!)
You will see
the following files:
- day1_exercise2_instructions.html this file!
- Si.scf.in this is an input file for scf calculations.
- bands.in this is an input file for
collecting bands.
- k-point-path this is a file containing list of
k-points along symmetry directions in Brillouin
zone.
- Si.plotband.in this is an input file for putting
band structure data into a plottable format.
- STEP 2:
Self-consistent (scf) calculations for Si
- Open and read the sample file Si.scf.in
- We chose values of celldm(1), ecutwfc, nk1,
nk2 and nk3 based upon our
results for exercise1. You may choose the same values as these, or
substitute your values.
- Run the scf
calculation:
/usr/local/apps/espresso-5.1/bin/pw.x < Si.scf.in > Si.scf.out
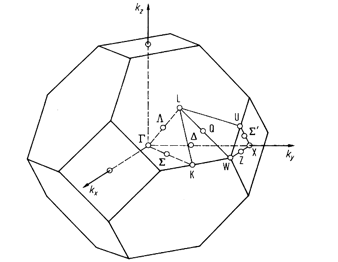
Figure 1: Brillouin zone of
Silicon (Diamond) structure
Band
structure of Si
-
- STEP 3:
Calculations for bands
- Copy
Si.scf.in to Si.band.in
- Edit Si.band.in
to perform non-self consistent calculations that will be used to obtain band
structure, by setting calculation='bands'.
- Add the parameter in &SYSTEM namelist called nbnd = 8
to specify the number of bands computed. Note that for a 2-atom Si cell,
we have 8 electrons and therefore only 4 occupied bands, but we are going
to compute some extra (empty) bands.
- Define the K_POINTS
card to specify the path along symmetry directions in Brillouin
zone. In order to this, you can use the information supplied in the file
called k-point-path, which
contains a list of k-points along high symmetry directions in the BZ,
i.e., L (½, ½, ½) to Gamma (0, 0, 0) to X (0, 0, 1) to W (0, ½, 1) to X
(0, 1, 1) to Gamma (0, 0, 0). You can paste that file to Si.band.in
after the K_POINTS card. (NOTE: You have change automatic to tpiba
which denotes that the k-points are in the units of 2p/a, and also remove the line “6 6 6 1 1 1”).
- Run a 'bands' calculation:
/usr/local/apps/espresso-5.1/bin/pw.x < Si.band.in > Si.band.out - What differences do you see
between the output file you obtain here, and the output file that you
obtained from your scf calculation?
- STEP 4: Collect
band results for plotting
- Open bands.in
- Note that you have to use the same
prefix in this calculation as was used in scf AND bands calculations.
- The flag filband defines the name of the file in which bands
data is to be stored.
- Run:
/usr/local/apps/espresso-5.1/bin/bands.x < bands.in > bands.out - Have a look at bands.out and bands.dat and
note the minimum and maximum energy eigenvalues at different k-points.
- STEP 5: Get the
data in a format to plot
- Open
Si.plotband.in
- The first line contains (a) input
file (= bands.dat obtained in the earlier step).
- Then (b) Emin and Emax
(= -6.00 and 10.00).
- (c) output
file in xmgrace format (bands.xmgr)
- (d) output
file in ps format (bands.ps)
- (e) Fermi energy
(= 6.337 eV)
- (f) deltaE and reference energy (= 1.00 6.337)
- Run the plotting
program:
/usr/local/apps/espresso-5.1/bin/plotband.x < Si.plotband.in > Si.plotband.out - You can view the plotted band
structure written in ps format (bands.ps)
using a postscript viewer (e.g., evince).