Group highlights
For a full list of publications see below or Google Scholar.
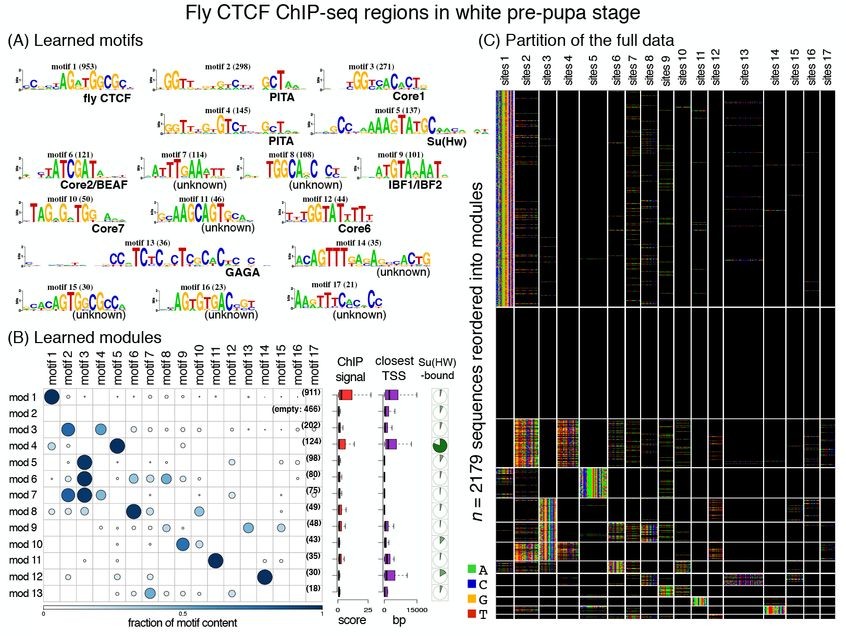
High-throughput sequencing-based assays measure different biochemical activities pertaining to gene regulation, genome-wide. The reported DNA regions are likely to be diverse, governed by different combinations of TF motifs. cisDIVERSITY is a new statistical framework, which models regions as modules characterized by distinct combinations of motifs. Both motifs and modules are learned de novo, directly from the data. It is therefore general enough to be applied to most high-throughput assays, such as ATAC-seq, ChIP-seq, GRO-seq, etc.
A Biswas, L Narlikar
Genome Research 31.9 (2021) 1646-1662
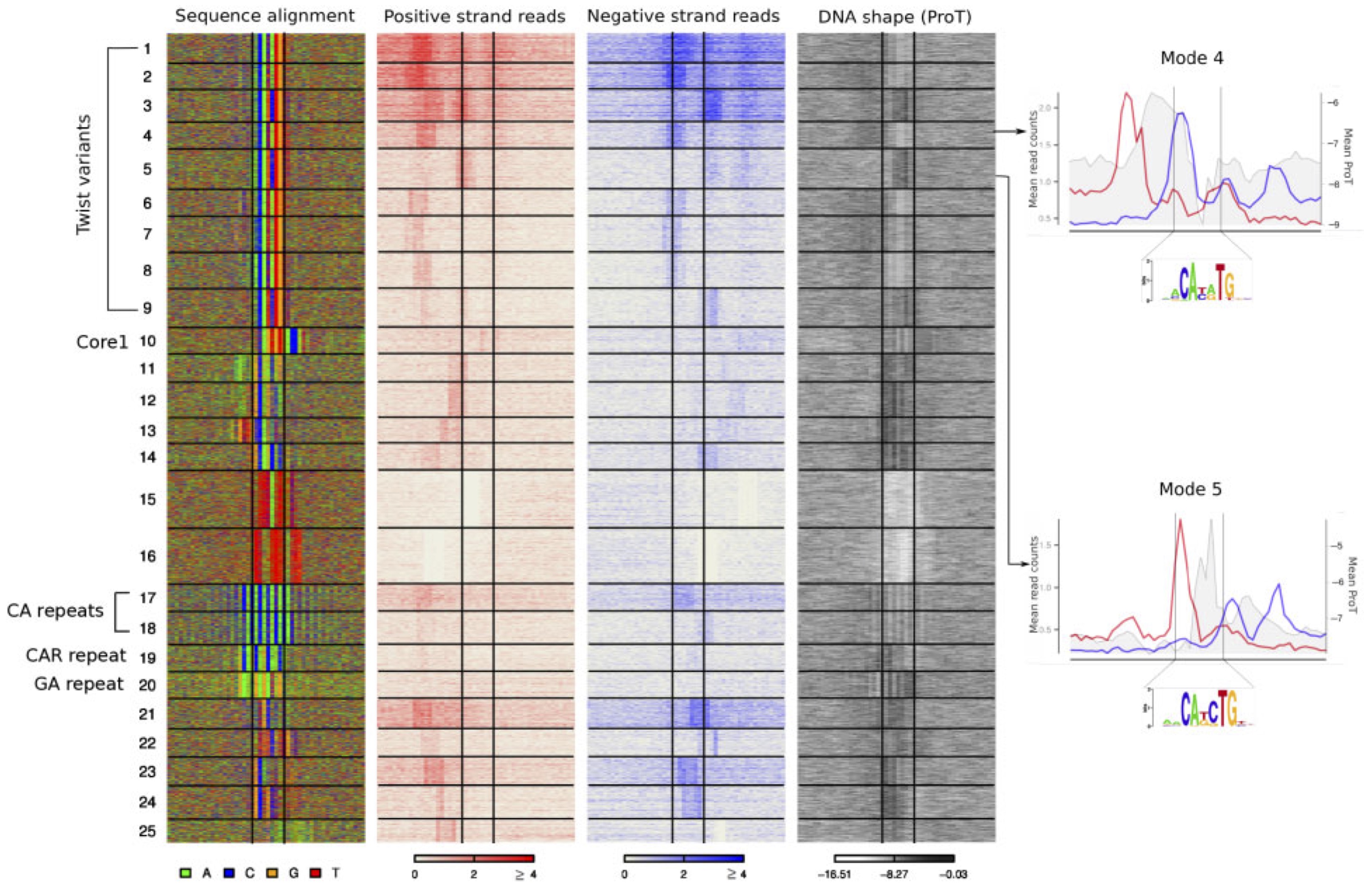
Exonuclease-based ChIP experiments improve the resolution of traditional ChIP experiments by cleaving DNA close to the protein-DNA complex. This results in a map of directional footprints on either side of the complex. These footprints are not necessarily identical at all locations: they depend on multiple factors, like the actual protein making DNA contact (which can be different from the target protein), the nucleotide sequence, and the precise location of the cross-link within the protein-DNA complex. We present an approach that resolves these footprints by learning a joint distribution over DNA regions and corresponding read distributions.
A Biswas, L Narlikar
Bioinformatics 37.S1 (2021) i367-i375
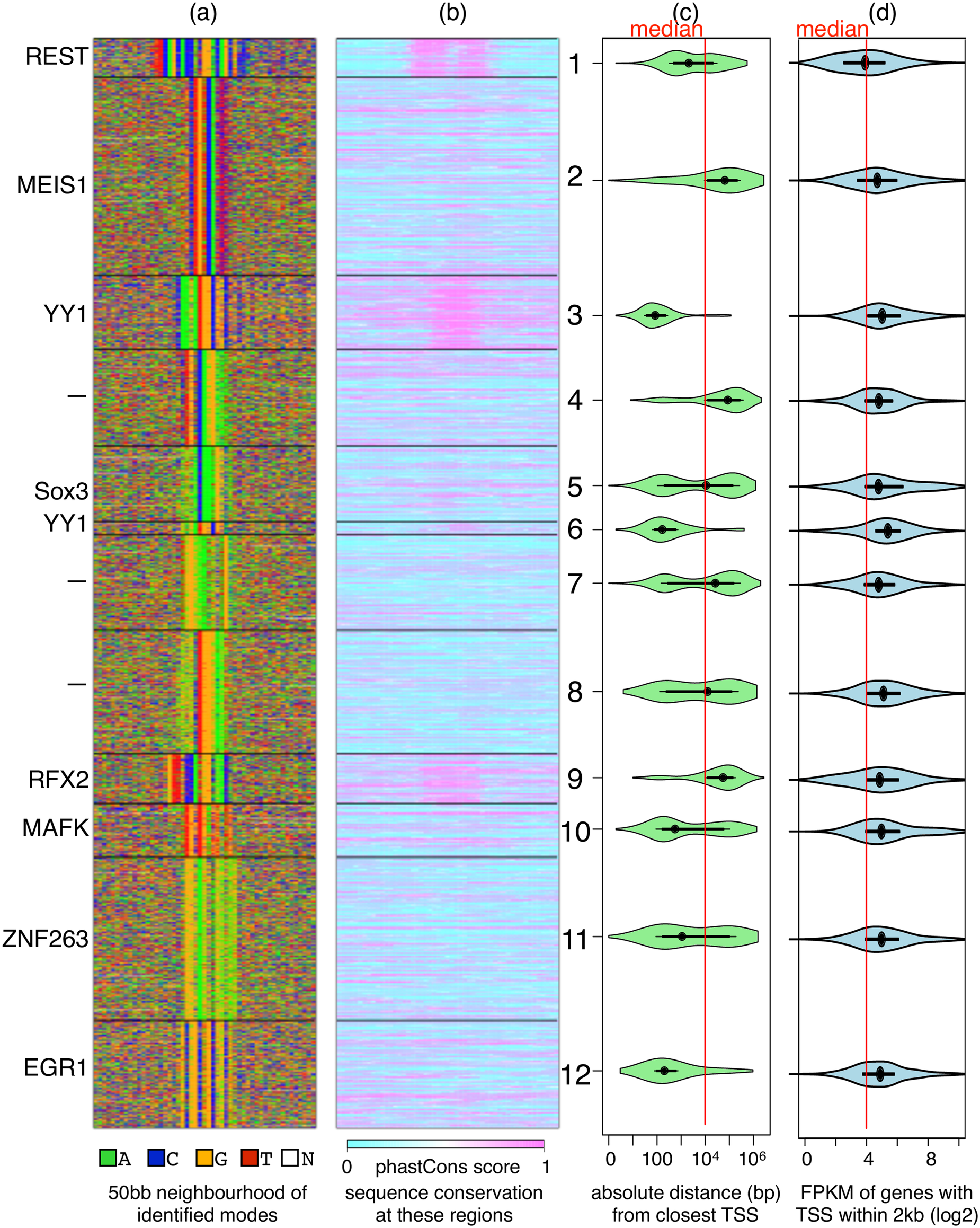
Our method Diversity models the fact that since a ChIP experiment pulls down regions participating in all complexes involving the profiled protein, the reported regions are in all likelihood, a collection of different types of protein-DNA contacts. DIVERSITY asks the question what sequence component caused a specific region to be reported in a ChIP experiment? The answer, in combination with additional data such as sequence conservation, SNPs, chromatin structure, downstream gene-expression, etc. yield insights into the diverse regulatory mechanisms at play.
S Mitra, A Biswas, L Narlikar
PLoS Computational Biology 14.4 (2018) e1006090
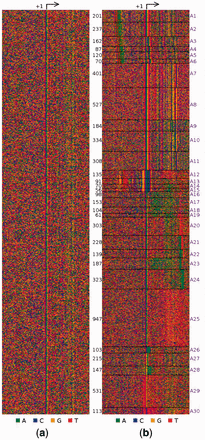
Promoters have diverse regulatory architectures and thus activate genes differently. For example, some have a TATA-box, many others do not. Even the ones with it can differ in its position relative to the transcription start site (TSS). No Promoter Left Behind (NPLB) is an efficient, organism-independent method for characterizing such diverse architectures directly from experimentally identified genome-wide TSSs, without relying on known promoter elements.
S Mitra, L Narlikar
Bioinformatics 32.5 (2016) : 779-781
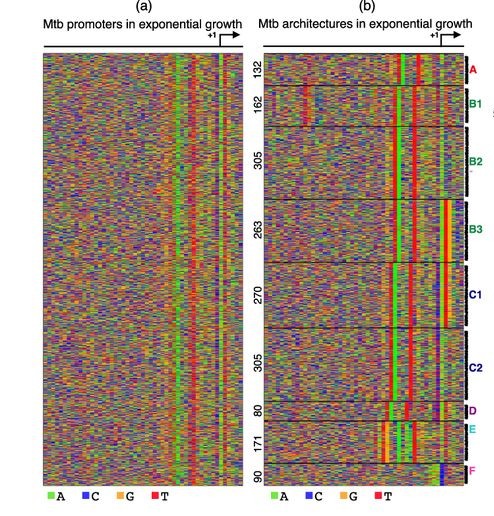
An important question in biology is how different promoter-architectures contribute to the diversity in regulation of transcription initiation. A step forward has been the production of genome-wide maps of transcription start sites (TSSs) using high-throughput sequencing. However, the subsequent step of characterizing promoters and their functions is still largely done on the basis of previously established promoter-elements like the TATA-box in eukaryotes or the -10 box in bacteria. We present a new, organism-independent approach that explicitly models this heterogeneity while unraveling different promoter-architectures.
L Narlikar
Nucleic Acids Research 42.20 (2014) 12388-12403
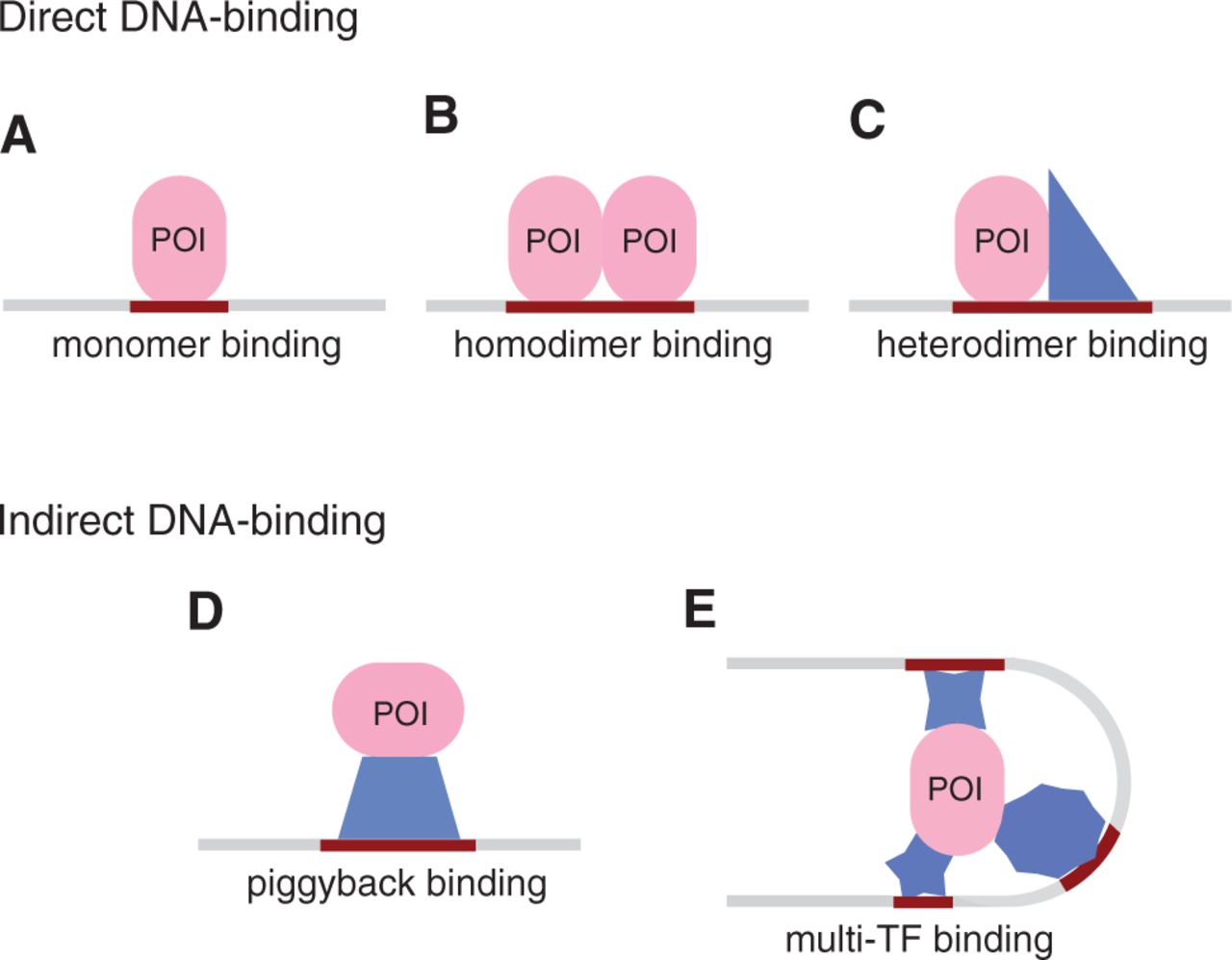
In chromatin immunoprecipitation-based high throughput experiments the protein binding to the DNA directly at its recognition site is not the only way the protein can cause the region to immunoprecipitate. Its binding specificity can change through association with different co-factors, it can bind DNA indirectly, through intermediaries, or even enforce its function through long-range chromosomal interactions. We present a novel Bayesian method that identifies distinct protein-DNA binding mechanisms without relying on any motif database.
L Narlikar
Nucleic Acids Research 41.1 (2013) 21-32
Full list
A fast method for clustering regulatory DNA regions
A Dileep, L Narlikar
IKDD CODS 2025 (to appear)
Accurate birth weight prediction from fetal biometry using the Gompertz model
C Kumari, GI Menon, L Narlikar, U Ram, R Siddharthan
European Journal of Obstetrics & Gynecology and Reproductive Biology X (2024) 100344
Ensemble learning for higher diagnostic precision in schizophrenia using peripheral blood gene expression profile
V Wagh, T Kottat, S Agrawal, S Purohit, T Pachpor, L Narlikar, V Paralikar, S Khare
Neuropsychiatric Disease and Treatment (2024) 20:923-936
Prediction of postpartum prediabetes by machine learning methods in women with gestational diabetes mellitus
D Parkhi, N Periyathambi, Y Ghebremichael-Weldeselassie, V Patel, N Sukumar, R Siddharthan, L Narlikar, P Saravanan
iScience (2023) 26:107846
Transcription factors organize into functional groups on the linear genome and in 3D chromatin
RN Vadnala, S Hannenhalli, L Narlikar, R Siddharthan
Heliyon 9.8 (2023) e18211
Machine learning prediction of non-attendance to postpartum glucose screening and subsequent risk of type 2 diabetes following gestational diabetes
N Periyathambi, D Parkhi, Y Ghebremichael-Weldeselassie, V Patel, N Sukumar, R Siddharthan, L Narlikar, P Saravanan
PLoS ONE 17.3 (2022) e0264648
Crowdsourcing assessment of maternal blood multi-omics for predicting gestational age and preterm birth
AL Tarca, BÁ Pataki, … , The DREAM Preterm Birth Prediction Challenge Consortium, JC Costello
Cell Reports Medicine 2.6 (2021) 100323
A universal framework for detecting cis-regulatory diversity in DNA regions
A Biswas, L Narlikar
Genome Research 31.9 (2021) 1646-1662
Resolving diverse protein-DNA footprints from exonuclease-based ChIP experiments
A Biswas, L Narlikar
Bioinformatics 37.S1 (2021) i367-i375
Orc4 spatiotemporally stabilizes centromeric chromatin
L Sreekumar, K Kumari, K Guin, A Bakshi, N Varshney, B C Thimmappa, L Narlikar, R Padinhateeri, R Siddharthan, K Sanyal
Genome Research 31.4 (2021) 607-621
MINDFUL: A Method to Identify Novel and Diverse Signals with Fast, Unsupervised Learning
M Parulekar, L Narlikar
bioRxiv (2019)
DIVERSITY in binding, regulation, and evolution revealed from high-throughput ChIP
S Mitra, A Biswas, L Narlikar
PLoS Computational Biology 14.4 (2018) e1006090
THiCweed: fast, sensitive detection of sequence features by clustering big datasets
A Agrawal, SV Sambare, L Narlikar, R Siddharthan
Nucleic Acids Research 46.5 (2018) e29-e29
No Promoter Left Behind (NPLB) : learn de novo promoter architectures from genome-wide transcription start sites
S Mitra, L Narlikar
Bioinformatics 32.5 (2016) : 779-781
Identification and computational analysis of gene regulatory elements
L Taher, L Narlikar, I Ovcharenko
Cold Spring Harbor Protocols 2015.1 (2015) pdb-top083642
Multiple novel promoter-architectures revealed by decoding the hidden heterogeneity within the genome
L Narlikar
Nucleic Acids Research 42.20 (2014) 12388-12403
One size does not fit all: On how Markov model order dictates performance of genomic sequence analyses
L Narlikar, N Mehta, S Galande, M Arjunwadkar
Nucleic Acids Research 41.3 (2013) 1416-1424
MuMoD: a Bayesian approach to detect multiple modes of protein-DNA binding from genome-wide ChIP data
L Narlikar
Nucleic Acids Research 41.1 (2013) 21-32
CLARE: Cracking the LAnguage of Regulatory Elements
L Taher, L Narlikar, I Ovcharenko
Bioinformatics 28.4 (2012) 581-583
ChIP-Seq data analysis: identification of Protein-DNA binding sites with SISSRs peak-finder
L Narlikar, R Jothi
Next Generation Microarray Bioinformatics. Humana Press (2012) 305-322
Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types
G Wei, B J Abraham, R Yagi, R Jothi, K Cui, S Sharma, L Narlikar, D L Northrup, Q Tang, W E Paul, J Zhu, K Zhao
Immunity 35.2 (2011) 299-311
Finding regulatory DNA motifs using alignment-free evolutionary conservation information
R Gordan, L Narlikar, AJ Hartemink
Nucleic Acids Research 38.6 (2010) e90-e90
Genome-wide discovery of human heart enhancers
L Narlikar, N J Sakabe, A A Blanski, F E Arimura, J M Westlund, M A Nobrega, I Ovcharenko
Genome Research 20.3 (2010) 381-392
Identifying regulatory elements in eukaryotic genomes
L Narlikar, I Ovcharenko
Briefings in Functional Genomics and Proteomics 8.4 (2009) 215-230
A fast, alignment-free, conservation-based method for transcription factor binding site discovery
R Gordan, L Narlikar, AJ Hartemink
Annual International Conference on Research in Computational Molecular Biology (RECOMB). Springer, Berlin, Heidelberg, 2008
Towards a complete transcriptional regulatory code: improved motif discovery using informative priors
L Narlikar
Dissertation Duke University, 2008
A nucleosome-guided map of transcription factor binding sites in yeast
L Narlikar, R Gordan, AJ Hartemink
PLoS Computational Biology 3.11 (2007) e215
Nucleosome occupancy information improves de novo motif discovery
L Narlikar, R Gordan, AJ Hartemink
Annual International Conference on Research in Computational Molecular Biology (RECOMB). Springer, Berlin, Heidelberg, 2007
Informative priors based on transcription factor structural class improve de novo motif discovery
L Narlikar, R Gordan, U Ohler, AJ Hartemink
Annual International Conference on Intelligent Systems for Molecular Biology (ISMB). Bioinformatics 22.14 (2006) e384-e392
Sequence features of DNA binding sites reveal structural class of associated transcription factor
L Narlikar, AJ Hartemink
Bioinformatics 22.2 (2006) 157-163